Binding evidence
The ligand should have a credible target engagement context, ideally supported by a structural pose and interpretable interaction pattern.
Viral PROTAC design is the process of asking whether a viral protein binder can become the target-binding arm of a bifunctional degrader. V-LiSEMOD supports that early decision by helping users review ligand pose, solvent-exposed atoms, exit-vector logic, target-side structural cues, and PROTACability evidence before deeper modeling or experimental work.
The goal of this page is not to claim that a degrader will work. It is to make the first design conversation more structured: which viral binders look chemically modifiable, structurally plausible, and worth follow-up?
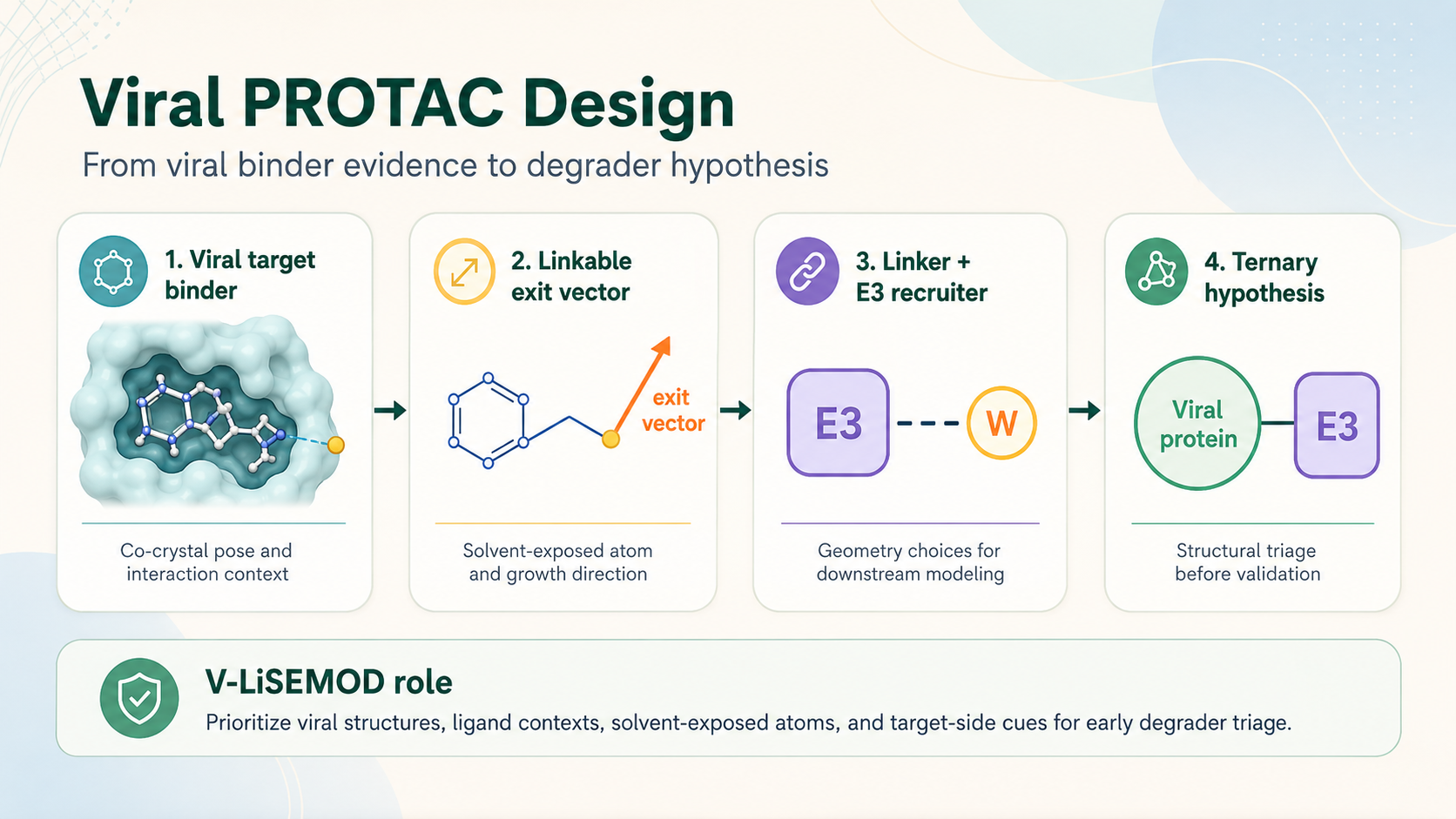
Antiviral degrader design is not simply a search for strong binders. A useful viral inhibitor must be evaluated as a possible warhead: it should bind the viral target, preserve important protein-ligand contacts after modification, and expose a chemical handle or moiety that could support a linker. V-LiSEMOD turns viral protein-ligand structural data into a review workflow for asking those questions in a consistent, evidence-driven way.
This page explains the design logic behind the viral PROTAC workflow, the types of evidence users should inspect, and how V-LiSEMOD can help prioritize structures for medicinal chemistry discussion, linker exploration, ternary complex modeling, or downstream experimental planning.
Viral proteins can have compact binding pockets, flexible loops, transient interfaces, conserved active sites, and context-dependent conformations. A ligand that works well as an inhibitor may still be a poor degrader warhead if the ligand is deeply buried, lacks a practical attachment point, or loses key interactions after chemical modification.
A degrader concept also has to do more than preserve viral target binding. It must support linker attachment, maintain a productive spatial relationship with an E3 ligase recruiter, and eventually create a cellular context in which target ubiquitination and degradation are possible. Those later steps cannot be confirmed from a co-crystal structure alone, but structural triage can identify which ideas are more reasonable to examine next.
In a PROTAC-style design concept, the viral ligand is often treated as the target-binding warhead. That label should be earned rather than assumed. A possible warhead should have an attachment strategy that is chemically reasonable and structurally aligned with the observed binding pose.
The ligand should have a credible target engagement context, ideally supported by a structural pose and interpretable interaction pattern.
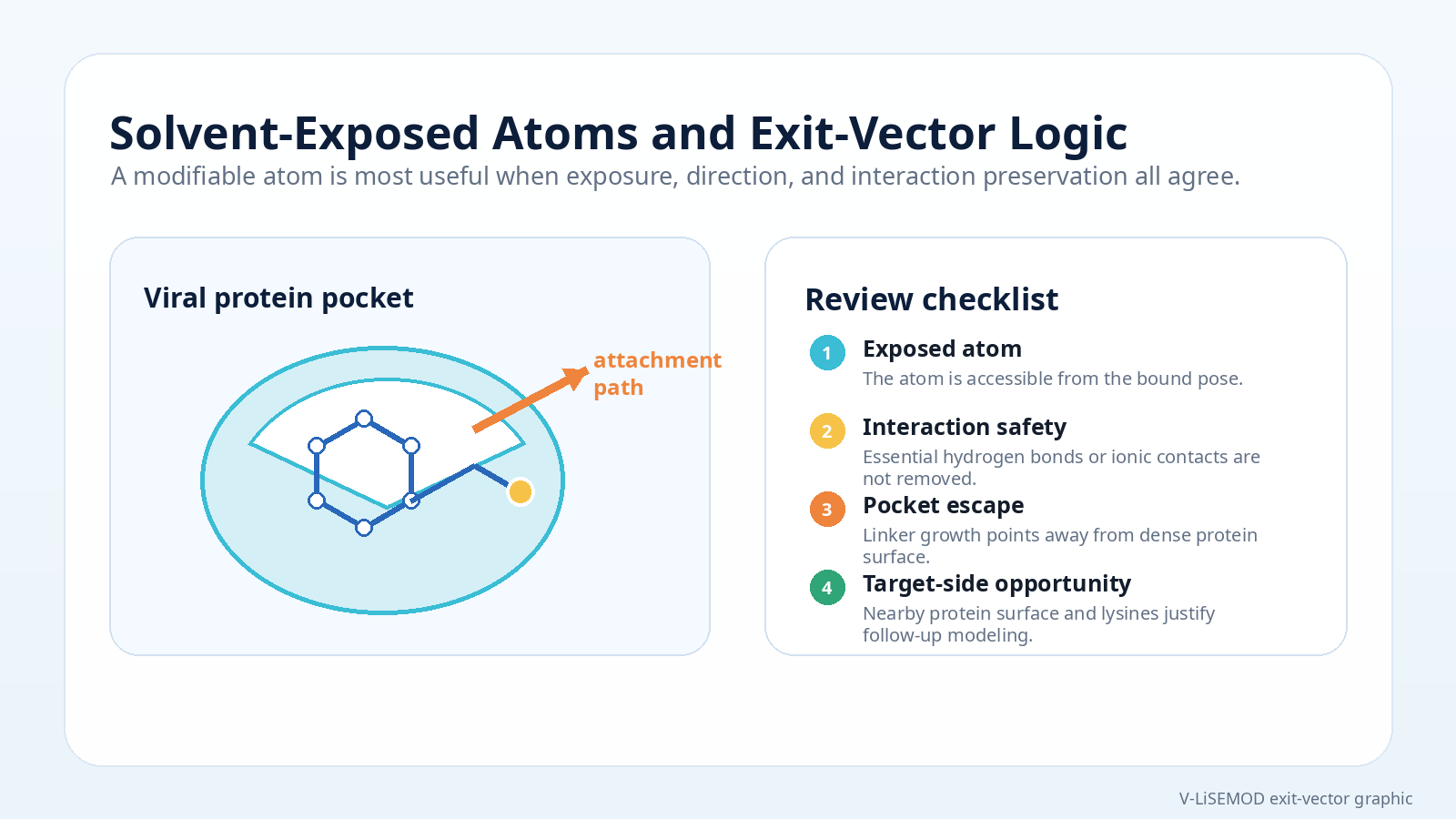
An outward-facing atom or region can suggest where linker growth may be less disruptive to binding.
The attachment point should point away from crowded pocket regions and toward space where a linker could plausibly extend.
Modification should avoid destroying interactions that appear central to binding, such as key hydrogen bonds, ionic contacts, or shape complementarity.
The most useful early workflow is not a single score. It is a sequence of questions that moves from target context to ligand pose, modification logic, linker planning, and follow-up evidence. V-LiSEMOD is designed to help users move through those questions without overstating what the structural data can prove.
Start with the protein, domain, pocket, chain, and viral system. A conserved catalytic pocket and a protein-protein interface can raise very different degrader design questions.
Review how the ligand sits in the binding site, which atoms are buried, and which interactions appear important for recognition.
Look for ligand atoms or moieties that face outward. These may become starting points for linkable-site discussion.
Ask whether the proposed attachment direction escapes the pocket and avoids obvious steric conflict with the viral target surface.
Use structural cues to decide which linker lengths, flexibility profiles, and E3 recruiters may be worth modeling.
Prioritized hypotheses still need medicinal chemistry, ternary modeling, cellular target engagement, degradation assays, and antiviral activity testing.

The observed pose helps define which atoms are buried, which interactions appear critical, and which regions may tolerate limited chemical growth.
Exposure mapping can highlight atoms or moieties that remain outward-facing after binding and therefore may be more compatible with modification.
Attachment is not just about exposure. The direction of growth matters for sterics, pocket escape, and downstream linker routing.
Chemical tractability, synthetic feasibility, and preservation of target engagement all matter when moving from binder to candidate warhead.
Accessible lysine context may support follow-up review, but it does not prove productive ubiquitination will occur.
Recruiter choice can shape geometry, permeability, selectivity, and feasibility, making downstream partner selection a major source of uncertainty.
Even promising structural cues cannot guarantee that a productive ternary complex will form or persist in cells.
Expression, localization, viral life-cycle stage, and host-cell biology can all influence whether a degrader concept has a realistic path forward.
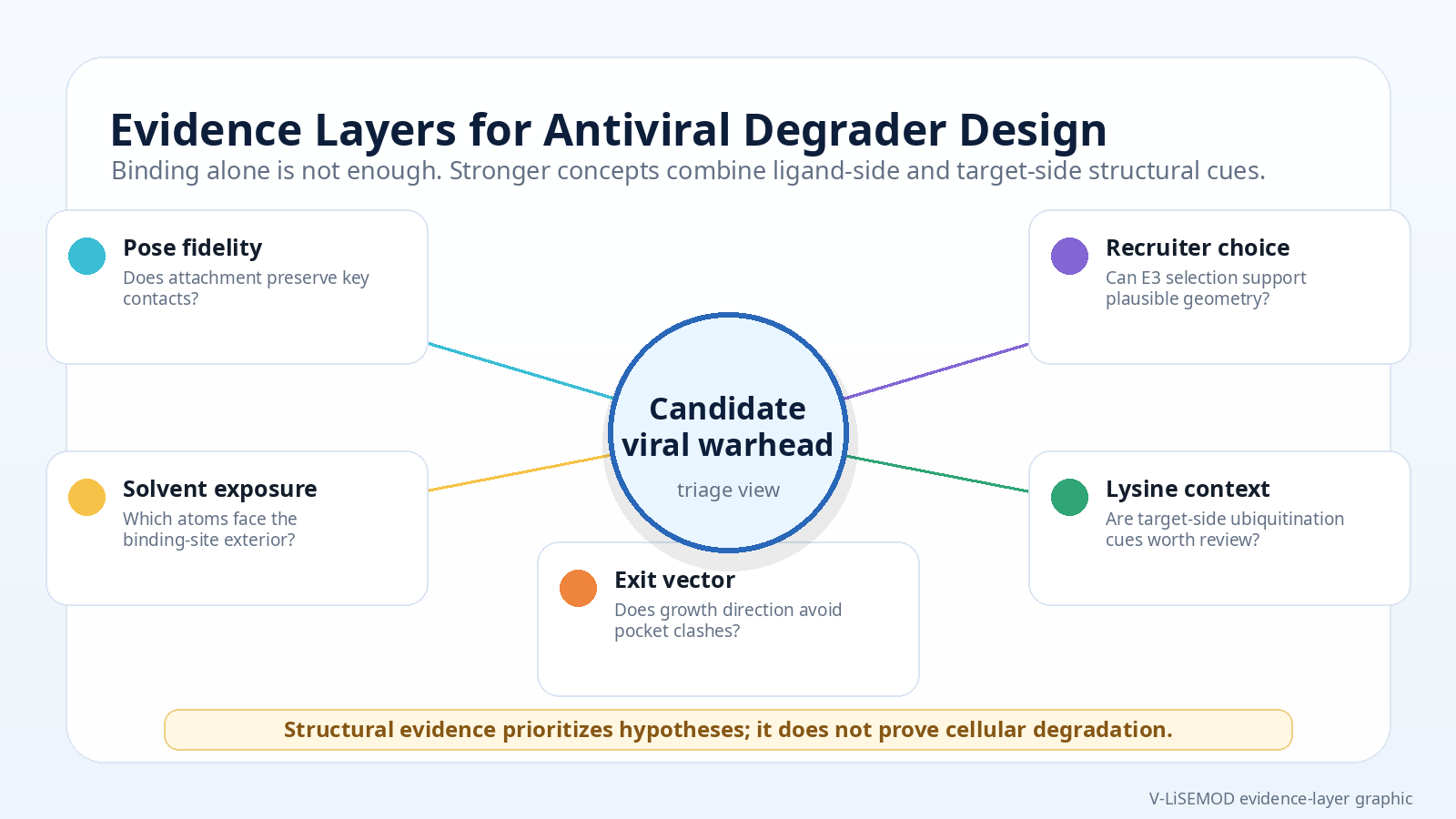
V-LiSEMOD helps prioritize viral structures for follow-up by combining ligand context, interaction evidence, solvent exposure, and target-side cues into a transparent review workflow. It is best used to narrow candidate structures and frame design conversations before medicinal chemistry, ternary modeling, or biological testing.
Compare structures to identify ligand poses with outward-facing atoms and interpretable protein-ligand interactions.
Separate ligands that may tolerate modification from binders that appear too buried or interaction-dependent.
Use exit-vector direction and pocket escape logic to decide which linker concepts are worth modeling first.
Use structural triage to support later PROTAC builder workflows, ternary complex modeling, or molecular visualization.
| Structural signal | Why it matters | How to use it |
|---|---|---|
| Outward-facing ligand atom | May indicate a possible linker attachment region. | Review whether modification would preserve the ligand pose and avoid major clashes. |
| Deeply buried ligand scaffold | May make linker attachment difficult without disrupting binding. | Treat as a caution signal unless a peripheral substituent remains exposed. |
| Critical polar interaction near proposed attachment site | Modification could remove or weaken an important recognition feature. | Prioritize alternative atoms, analogs, or attachment positions. |
| Accessible target surface near ligand exit path | Can support follow-up review of ternary complex geometry. | Use as a modeling prompt, not as proof of degradation. |
| Candidate target lysines | Lysine context can matter for ubiquitination hypotheses. | Review residue accessibility, orientation, and cellular context before overinterpreting. |
V-LiSEMOD is a prioritization and review aid. It does not claim that a ligand will become a successful antiviral PROTAC, that an E3 recruiter will form a productive ternary complex, or that the viral target will be degraded in cells.
It is the effort to adapt a viral protein binder into a degrader concept by connecting the target-binding ligand to an E3 ligase recruiter through a linker.
In this context, PROTACability means the structural plausibility that a viral target-ligand system may support degrader design review. It is not the same as proven degradation.
They can reveal ligand regions that face outward from the binding site, which may be more compatible with linker attachment than buried atoms.
Not necessarily. A strong inhibitor can still be unsuitable if it lacks a modifiable exit vector, loses binding after attachment, or fails to form productive degrader geometry.
No. V-LiSEMOD helps users prioritize structures and hypotheses for follow-up. Degradation must be tested with appropriate experimental assays.
Use V-LiSEMOD first to reason about candidate viral warheads and attachment points, then use builder workflows to explore linker and recruiter combinations for downstream modeling.
Use the PROTACability workflow when you want to inspect viral protein-ligand structures, compare candidate binders, and decide which antiviral degrader hypotheses deserve deeper attention.